

研究の概要
背景と課題
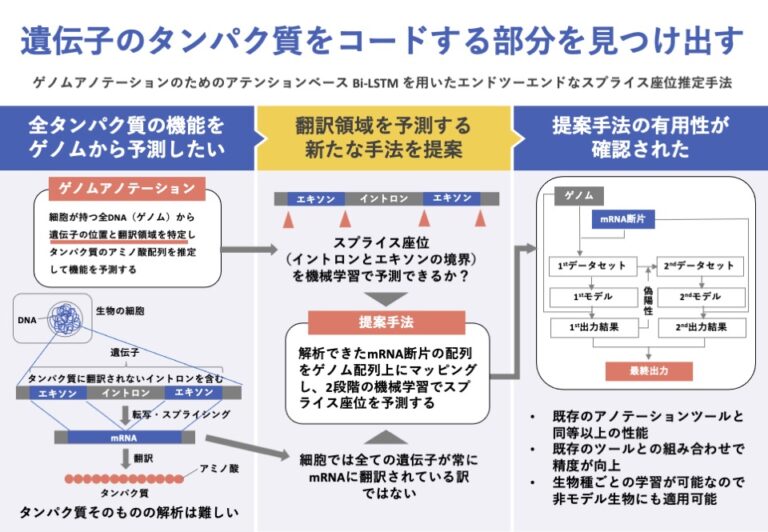
生物の遺伝情報はDNAにコード化されて保存されており、この情報を遺伝子と呼ぶ。タンパク質のアミノ酸配列をコードする遺伝子はRNAの一種であるmRNAに転写され、mRNAはタンパク質に翻訳され、発現する。
タンパク質はアミノ酸が線状に連なった高分子ポリマーであり、細胞内でのさまざまな生命活動を担う。アミノ酸配列の類似性からタンパク質の機能をある程度予測することができるため、ゲノム上にコードされているタンパク質のアミノ酸配列を決定することは、様々な生物学的解析の基礎となる。そのため、新規ゲノムを決定した際に行なわれるのがゲノムアノテーションである。
ゲノムアノテーションは2段階に分かれる。まず、構造アノテーションでは、遺伝子の位置とコード領域を特定し、その遺伝子がコードする全タンパク質配列を推定する。そして、機能アノテーションでは、得られたタンパク質配列の機能を、配列の類似性などに基づいて予測する。
真核生物ゲノムの遺伝子には、タンパク質をコードする部分(エキソン)とそうでない部分(イントロン)がある。そのため、真核生物ゲノムの構造アノテーションではエキソンとイントロンの境界であるスプライス座位(SS)を特定する必要がある(図1)。
一般的に、SSを特定する方法はRNA-seqデータ(次世代シーケンサーを用いて解読したmRNA断片配列)をゲノムにマッピングすることである。しかし、一部の遺伝子は発現が乏しい、あるいは非定常であり、取得したRNA-seqデータで全ゲノムをカバーすることは困難である。
本研究では、対象生物種のゲノムとRNA-seqデータを入力とし、トレーニングデータの作成、モデルの学習を自動的に行い、そのゲノムに特化した予測モデルを作成することで、SSを推定する手法を提案する。
提案手法
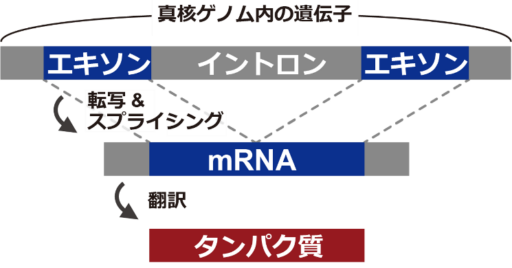
ゲノム上ではスプライス座位(SS)の数が少なく、ほとんどの座位が非SSである、という不均衡データ問題を緩和するために、提案手法ではデータセットを構築、学習、予測を2段階で行う(図2)。
提案手法は、次の8ステップから構成される。
- HISAT2(Kim et al. 2019)と SAMtools(Li et al. 2009, Li 2011)を用いて、RNA断片をゲノム上にマッピングすることで明らかなSSと非SSを識別し、SSと判定されたtrueデータと非SSと判定されたfalseデータで構成される第1データセットを作成する。
- 第1データセットを用いて第1モデルを学習させる。
- 第1モデルを用いて、RNA断片がカバーする座位を含むゲノムの全座位を予測する。
- データの不均衡問題に起因するFP(偽陽性)を抑制するため、RNA断片がカバーするゲノム領域(確実にSSである座位が分かっている)に基づいて、第1モデルの予測値を検証する。ここで特定されたFPは第2データセットに利用される。
- 第1データセットと同じtrueデータと、前ステップで特定されたFPを用いて、第2データセットを作成する。
- 第2データセットを用いて第2モデルを学習させる。
- 第2モデルを用いて、ゲノムの全座位を予測する。
- 第1モデルと第2モデルの両方からSSと予測された座位を特定し、最終結果として出力する。
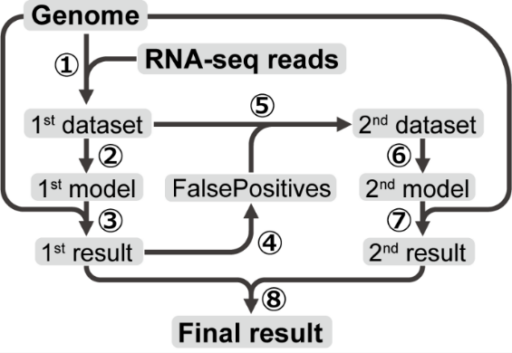
提案手法は2つの分類器(モデル)を学習させる。それぞれのニューラルネットワーク(NN)モデルは、Bi-LSTM(双方向長短期記憶)層、アテンション層、全結合層の3層で構成される(図3)。モデルへの入力は固定長のDNA配列で、最終的な出力はSoftMax活性化関数で正規化された、入力配列の中心座位がSSであるかどうかの確率である。
実験
性能評価のための実験を行った。固定量の短いDNA配列で構成されたデータセットを用いたモデル構造の比較実験と可変長の長いゲノム配列を用いた検証実験を行った。後者の実験では、既存の代表的な手法との性能比較も行った。
データセットを用いた比較実験では、混同行列を作成し、F1スコアを評価指標とした。長いゲノムを用いた検証実験では、SSをtrue、それ以外の座位をfalseとして、ゲノム上の全座位を1つのデータとしてカウントし、混同行列を作成し、適合率、再現率、F1スコア、マシューズ相関係数(MCC)を算出した。
固定長配列データセットを用いたモデル構造に関する実験
提案するモデル構築の妥当性を評価するために、学習に十分な数のイントロンを持っている、H. sapiens(ヒト)、A. nidulans(カビ)、K. bialata(海洋微好気性単細胞生物)のゲノムを選択した。
まず、H. sapiens(NC_000002.12)、A. nidulans(GCF_000149205.2)、K. bialata(GCA_003568945.1)のゲノムとアノテーションデータから第1、第2データセットを構築した。データセットのラベル分布は、H. sapiensでは両データセットともに29,695のtrueと118,780のfalse、A. nidulansでは両データセットともに48,496のtrueと193,984のfalse、K. bialataでは第1データセットでは10万のtrueと40万のfalse、第2データセットでは10万のtrueと156,545のfalseがあった。
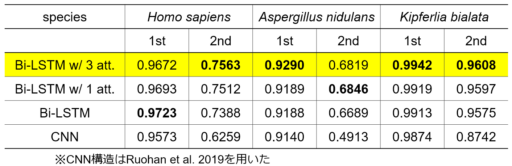
これらのデータセットに対して5分割交差検証を行い、本モデルと複数のモデル構造で性能(F1スコア)を比較した。比較対象として、Bi-LSTM w/o attention、single-attention Bi-LSTM、CNNを選択した。
その結果、どのモデル構造も第1データセットでは高い性能を発揮したが、第2データセットでは第1データセットより低いスコアとなった(表1)。提案したモデル構造は、総合的に最も優れた性能を達成した。
長いゲノムを用いた検証実験
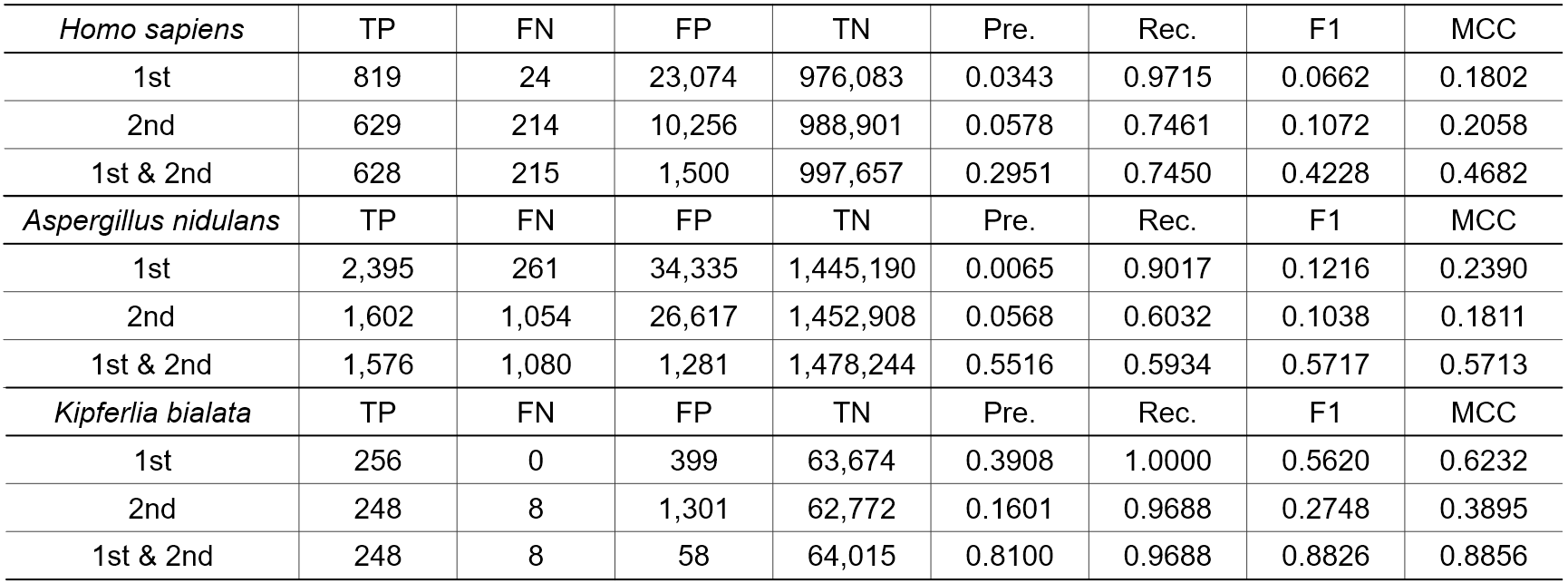
次に、固定長配列データセットではなく、可変長の長いゲノムを用いて実験を行った。3生物種のゲノムから、検証のために部分的なゲノム断片を除外し、提案手法の学習を行った。トレーニングは3つの種について独立して行われ、検証のために除外された各ゲノム断片のSSを予測した。
提案手法には2つのモデルが含まれるので、それぞれのモデルの検証結果と、2つのモデルの論理積をとった最終的な出力の結果を示す(表2)。両モデルとも、単独ではFPが高く、精度も非常に低かった。しかし、両モデルを組み合わせることで、FPは大幅に減少し、F1スコアとMCCは増加した。
既存手法との比較・統合
比較するツールとして、様々なアノテーションパイプラインの中で最もよく使われているGeneMarkと、複数のアノテーションツールを組み合わせて使うパイプラインツールであるFunannotate(Palmer & Stajich, 2020)を選択した。GeneMarkについては、ESモード(Ter-Hovhannisyan et al. 2008)と、ETモード(Lomsadze et al. 2014)をテストした。
選択したツールによるアノテーション結果からSSの位置のみを抽出し、提案手法と同様に評価指標を算出した。また、提案手法と各既存ツールの結果との論理積のスコアも算出した。
その結果、H. sapiensとK. bialataでは、Funannotateと提案手法の組み合わせが最も適合率、F1スコア、MCCが高くなった。また、この2種では、提案手法が最も高い再現率を記録した。一方、A. nidulansでは、GeneMark-ETが最も高い適合率、F1スコア、MCCを示し、GeneMark-ESと提案手法の組み合わせが最も高い再現率を記録した。3種とも、提案手法と従来のツールを組み合わせることで、FPが減少し、適合率が上昇した。また、H. sapiensとK. bialataでは、F1スコアとMCCが増加した。
議論と結論
本研究では、アテンションベースのBi-LSTMに基づくモデルを用いて、ゲノムとRNA-seqデータからスプライス座位(SS)を学習・予測する新規ゲノムアノテーションに適用可能な手法を提案した。
これまでのニューラルネットワークに基づくSS予測手法の多くは、ヒトなど既にアノテーションされたモデル生物のゲノムに対して事前にトレーニングすることで最適化されていた。提案手法は、生物種ごとにトレーニングできるため、非モデル生物の新規ゲノム配列のアノテーションに有効である。さらに、提案手法は、異なるデータセットで学習させた2つのモデルの結果を組み合わせることで、不均衡データ問題に対処している。
これまで提案されてきたアノテーションツールと比較して、提案手法は同等の性能を示し、他の手法と組み合わせることで精度を向上させることができた。これは、提案手法が、より質の高い遺伝子モデルの提供に貢献できることを示しており、さらなる生物学的解析に有用であることを示している。